General Statistics of DIONYSUS
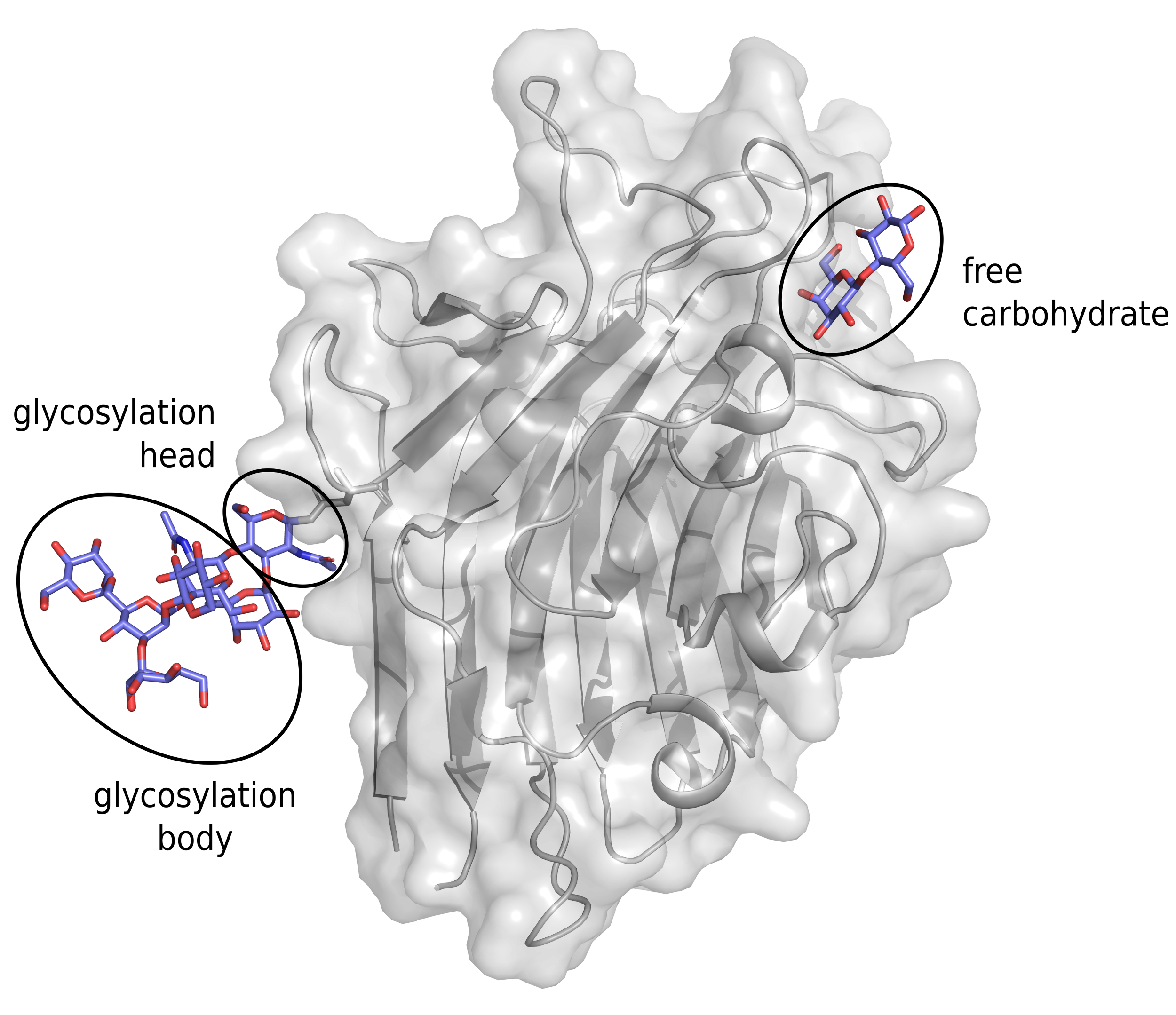
Protein-Carbohydrate Interfaces
We distinguish three types of interfaces between protein and carbohydrate: free carbohydrate (strictly non-covalent interaction), glycosylation heads (a sugar moiety covalently bound to a protein residue) and glycosylation body (a part of carbohydrate covalently bound to the glycosylation head and eventually interacting with the protein residues non-covalently).
Here are some statistics on the database content in terms of different protein-carbohydrate interfaces:
Carbohydrate Residues
Here is the content in terms of different carbohydrate components:
Proportion of Protein Functions
Here is the proportion of different protein functions according to different databases. To eliminate some structural redundancy, we only consider one protein per Uniprot ID
Proportion of Organisms
Here is the proportion of different protein classes:
Protein Structure Page
Chains overview

Individual chain properties

Binding site visualization

Related structures



Alignment and Score
In order to obtain comparable sizes of interfaces, each sugar binding site was defined as residues in the proximity of one carbohydrate moiety. Therefore, for a pentasaccharide molecule we have extracted five different binding sites. Each binding site was prepared as follows:
- For a given sugar residue, select its carbohydrate ring
- Select all protein atoms, which are located at closer than 7 Å from any atom of the carbohydrate ring and located on the protein surface (solvent accessibility surface area $\gt 0$)
- For all the selected protein atoms we add into consideration $C_\alpha$ of the corresponding residue (in order to improve robustness of the following calculations)
Then, we have applied the following algorithm to compare two structures $\mathcal{S}_1$ and $\mathcal{S}_2$.
For each atom of the binding site (carbohydrate ring and protein atoms selected as described above) we assign an atom type with respect to their physico-chemical properties. For protein atoms, we assign types according to the residue and atom names (e.g. distinguishing $C_\alpha$ atoms from aromatic carbons). In order to avoid mapping between protein and carbohydrate atoms, we assign specific atom types for carbohydrate rings.
We consider all pairs $i$ and $j$ of corresponding atoms. A correspondence between two atoms exists only if they are of the same type. Let’s denote $i=(i_1, i_2)$ pairs of atoms of the target protein and pairs $j=(j_1, j_2)$ of the carbohydrate ligand.
The distortion between atoms $i_1$ and $j_1$ and atoms $i_2$ and $j_2$ is:
$\Delta_{ij} = |(\text{dist}(i_1, j_1) - \text{dist}(i_2, j_2))|$
where dist($i_1$, $j_1$) is the euclidean distance between atoms $i_1$ and $j_1$
A matching between $\mathcal{S}_1$ and $\mathcal{S}_2$ denoted by $M$ consists in a set of atom pairs $(i_1, i_2)$, $i_1 \in \mathcal{S}_1$ and $i_2 \in \mathcal{S}_2$, where $M$ gives a one-to-one mapping between $\mathcal{S}_1$ and $\mathcal{S}_2$. The mean distortion is:
$MD(\mathcal{S}_1, \mathcal{S}_2) = \displaystyle \frac{1}{|M|^2}\sum_{i,j}|\text{dist}(i_1, j_1) - \text{dist}(i_2, j_2)|$
The comparison algorithm aims to compute the largest matching such that the mean distortion between structures is below a given threshold noted ∆max. This objective is satisfied by solving the following optimization problem:
$\displaystyle \max_M|M|$ such that $MD(\mathcal{S}_1, \mathcal{S}_2) \leq \Delta_{\max}$
For this purpose, a maximum clique approach is applied to the correspondence graph. Nodes of the correspondence graph are pairs $i=(i_1, i_2)$ and an edge connects node $i$ to node $j$ if $i_1 \neq i_2$ and $j_1 \neq j_2$ and the distortion $\Delta_{ij}$ is less than max fixed to 1 Å.
References:
- Rasolohery I., Moroy G., Guyon F. "PatchSearch. A Fast Computational Method for Off-Target Detection."" J. Chem. Inf. Model. (2017)
- Rey, J., Rasolohery, I., Tufféry, P., Guyon, F., & Moroy, G. "PatchSearch: a web server for off-target protein identification." Nucleic Acids Res. 47 (2019)
- Patchsearch webserver
- Moroy G., Guyon F. "An efficient non-sequential alignment of binding sites for fast peptide screening." (submitted)