
A short explanation of the Protein Blocks

The Protein Blocks.
Why a structural alphabet ?
The study of the local conformations of protein structures had a long history principally based on the study of the classical repetitive structures (alpha-helix and beta-sheet) and some characterizations of the coil state (turns). The secondary structures are interesting to describe the global fold protein, but does not take into account the specificity of the coil and begin/end of repetitive structures.
The structural alphabet : two goals.
Our work had consisted in (i) building a set of Protein Blocks able to approximate at best the different structural patterns observed along the protein backbones, and (ii) in predicting the local 3D-structure of the backbone in terms of PBs from the knowledge of the sequence.
Methods.
Identification of the different protein blocks is performed by an unsupervised cluster analyser taking account the sequential dependence of the blocks. After a phase of training performed on a given non-redundant protein databank, the prediction of the Protein Blocks from the knowledge of the protein sequence by Bayes theorem is done.
The Information.
Proteins are selected in a databank of non-homologous protein structures. Each protein backbone is translated into series of the dihedral angles (phi, psi).
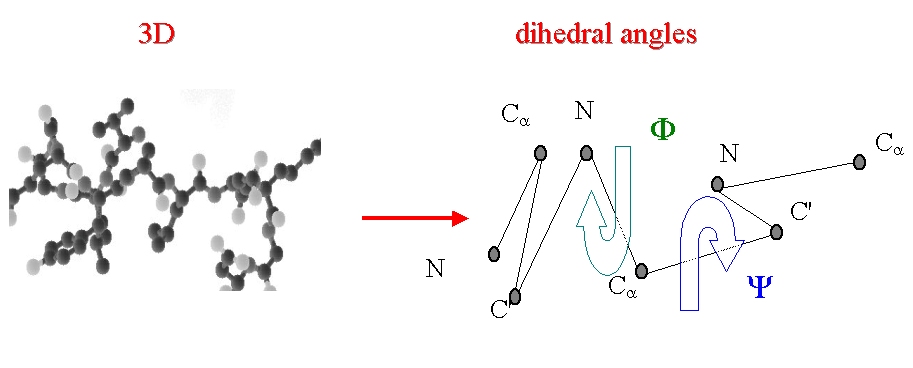
Description of the protein backbone in terms of dihedral angles Phi and Psi.
For the analysis, the proteins are splitting up in fragments of 5 consecutive residues to define the Protein Blocks. Two set of proteins have been defined one for the training stage, another for the the stage of prediction accuracy assessing. The fragments used are overlapping. Hence, a protein of length L is described by L-4 fragments. The goal is to define a structural alphabet for coding the local 3D structure of protein backbones. This alphabet is composed of Proteins Blocks" (PBs) which represent average patterns of the backbone fragments extracted from the databank.The learning method uses the principle of the self-organized learning of the Kohonen network, by reading a certain number of times, the totality of the vector databank in order to define the mean weights of the neurons. In practice, we have defined B Protein Blocks (B=34 at the begining), by taking randomly B fragments of 5 Calpha (=8 angles) in the databank.
Learning method.
1- A protein fragment of 8 consecutive dihedral angles (called "dihedral vector") is taken randomly in the learning databank.
2- This dihedral vector is compared to the B PBs, to define the closest PB. The dissimilarity measure between a fragment and a PB is defined as the Euclidean distance among the 8 angles, and is called RMSda for Root Mean Square deviations on angular values.
3- The closest PB from the fragment studied is obtained through the minimal rmsda value.
4- Then, the chosen PB is slightly modified to learn this fragment. An other fragment is taken and the process repeats from step one.
The training is iterative: a certain number C of cycles of readings of the vector databank is needed for defining the optimal Protein Blocks.
A second step similar to Hidden Markov Model of order 1 is then performed. So, at the end of the process, each PB is represented by an average dihedral vector. B PBs are finally obtained. We have tested different series of B PBs, with B = 34, 22, 19, 18, 16, 14, 12, 11 and 10. The serie with B=16 PBs has been chosen as the best correspondence between a good structural approximation and a correct local prediction.
some data:
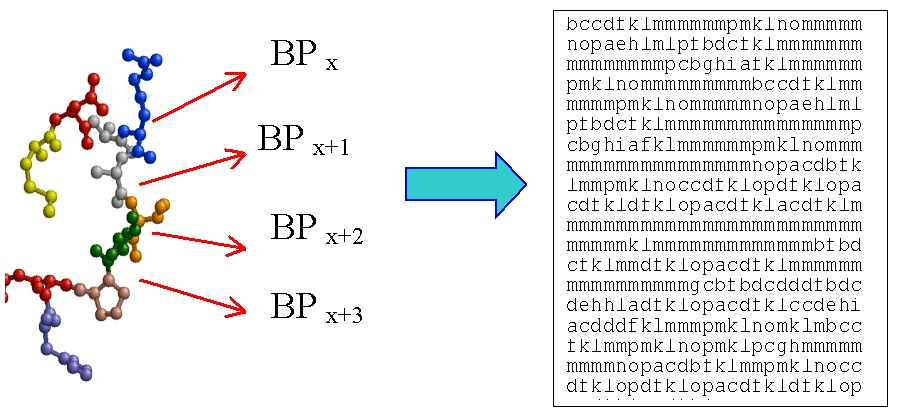
A protein structure could be coded as a succession of Protein Blocks.
Prediction.
After this phase of training performed on a given non-redundant protein databank, we can tackle the problem of the prediction of these structural blocks from the knowledge of the protein sequence. From a given library of PBs, amino acid preferences for different positions along the fragment can be extracted for each fold pattern. Using Bayes theorem, these probabilities may be further used to predict the structural motifs able to be adopted by a given protein chain. In our study, we have worked in different aspects to improve the PB prediction : (i) {1 protein block - n sequences}: Associating one protein block with one class of sequences is a restrictive point of view. A same fold pattern (or PB) may be associated with different types of sequences. We have defined a new procedure to split the set of fragment sequences associated with one PB into different clusters. These clusters allow a better description of the sequence specificities associated with each PB. These cluters are called "sequence families". (ii) {1 sequence - n protein blocks}: It is the inverse concept. Similar sequences are not always associated with the same fold, but with different "possible" folds. So we can devise a "fuzzy model" in which we have a certain probability for finding the true PB (this one that approximates at best the local structure of the backbone) among the proposed PBs. Concerning the existence of this "fuzzy model", we ought to check that the true PB is present among the solutions of the r first ranks (i.e. having the best prediction scores) provided by the Bayesian approach. We have defined an entropy-based index. It is used to discriminate different zones of the protein with high probabilities of prediction. So, two main directions have been explored. The first one called "global strategy" consists of locally determining the optimal number of protein blocks to be selected after fixing the prediction rate for the whole protein sites. So, the number of selected solutions per position may be variable. In contrast, the second direction called "local strategy" scans the protein sequence with a fixed number of solutions (i.e. a constant number of protein blocks per position) and determines the regions able to be predicted with this given number and with a fixed prediction accuracy. : The Protein Blocks Prediction Server.
: The Protein Blocks Prediction Server.
Some rates.
The initial prediction rate was 30.0% and is based on the simple use of 5 amino acid length sequence, i.e. the length of the PB. The increase of the length to 15 consecutive residues increase the prediction rate to 34.4%. Indeed, the information contains in the sequence is higher than expected. The true PB is found with a 71.5% rate within the 4 most probable PBs. The sequence families permit to increase the prediction rate to 40.7% and now more than 75% of the true PB are found within the 4 most probable PBs. The main point of the sequence families is the decreasing of the difference between the PB with the highest prediction rate and the one with the lowest. back