Help
What can you find in DIONYSUS?
- Structures: This table contains properties associated with individual Protein Data Bank (PDB) entries. For a full list of structures available, please visit this page.
- Proteins: Each row in this table corresponds to a distinct protein, defined by its specific entity within the PDB database. Additionally, you can explore glycosylation details of these proteins here.
- Binding site: As the central aspect of DIONYSUS, each entry corresponding to a particular binding sites. Special effort has been made to collect all binding sites, including breaking down components representing polysaccharides into their individual sugar rings, capturing one site per model, and accounting for each site with alternate locations when multiple exist. Users can search, compare, and analyze clustering of these binding sites through the following features: Detailed tutorials exists on this page to help users navigate through these tools.
- Component: This table provides information about each distinct component within the database. A full list of components can be accessed here.
- In addition to these core features, DIONYSUS also offers users the ability to inspect specific structures by navigating to https://www.dsimb.inserm.fr/DIONYSUS/structure/pdb_code. For instance: 1DIW.

Browse sugar-binding database
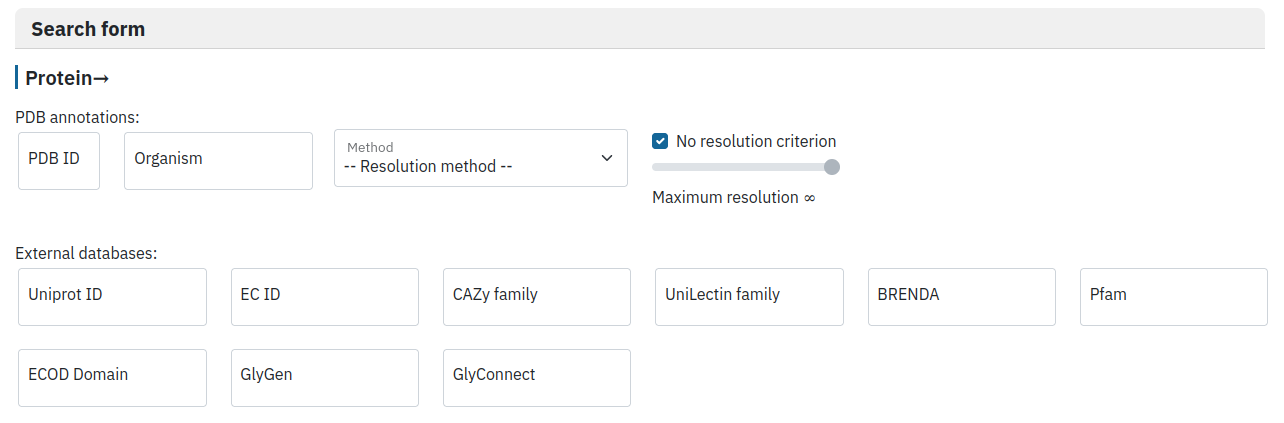
Filter by protein properties
The user is invited to provide a protein ID in any of the listed databases, its organism of origin or maximal resolution (for X-ray and EM structures).
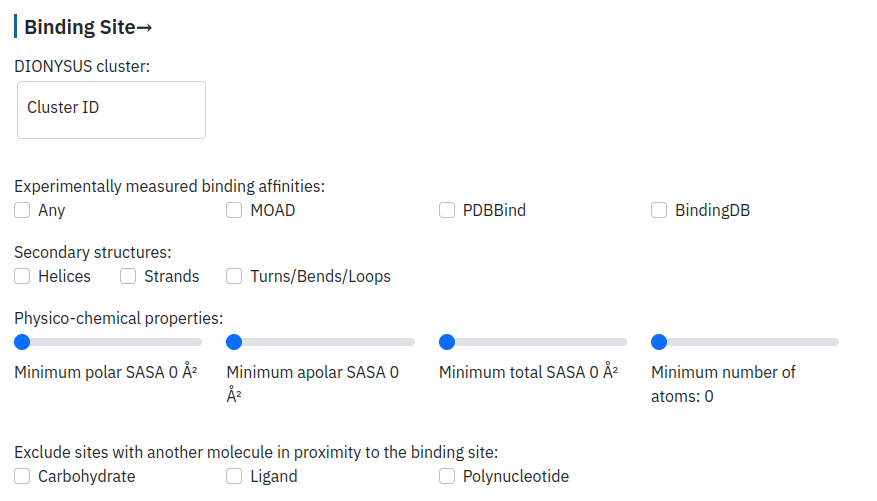
Filter by the binding site properties
The user can provide a cluster ID ("Explore clusters of sugar binding sites" section) and specify if she/he would like to keep only the entries with available affinity values. The user can provide desired specifications on the structure and composition of the binding site, such as its content in terms of secondary structure and solvent accessible surface area (SASA) for the whole binding site or for the polar/non-polar amino acids. Finally, the user can eliminate from consideration all the entries that contain another residue in the direct proximity of the carbohydrate ligand: carbohydrate (as a separate molecule or another carbohydrate moiety in polysaccharides), another ligand (small molecule, peptide, lipid, or ion), or polynucleotide ligand.
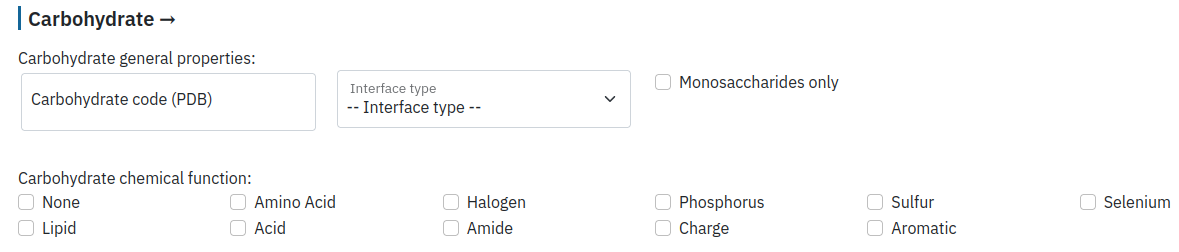
Filter by carbohydrate properties
The user can select a particular residue name (for example, GLC) as well as the interface type. We distinguish three types of interfaces between protein and carbohydrate: free carbohydrate (strictly non-covalent interaction), glycosylation heads (a sugar moiety covalently bound to a protein residue), and glycosylation body (a part of carbohydrate covalently bound to the glycosylation head and eventually interacting with the protein residues non-covalently). See "About" for further details.
The user can select only carbohydrate monomers (monosaccharides) as well as its chemical function.
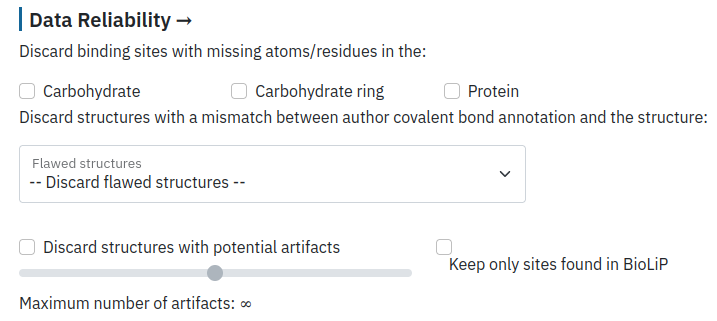
Filter by data reliability
PDB data on carbohydrates contain a lot of artifacts, such as missing atoms/residues both in protein and carbohydrate structures, which can be filtered during this step. The user can also filter binding sites containing mismatches between the author annotation and distances retrieved directly from the PDB, such as structures with abnormal glycosylation bond (annotated as glycosylation by authors) and binding sites which correspond to glycosylation sites but are not annotated as such by the authors. Finally, the user can select only interactions annotated in the BioLip database as biologically relevant to further refine the search results.
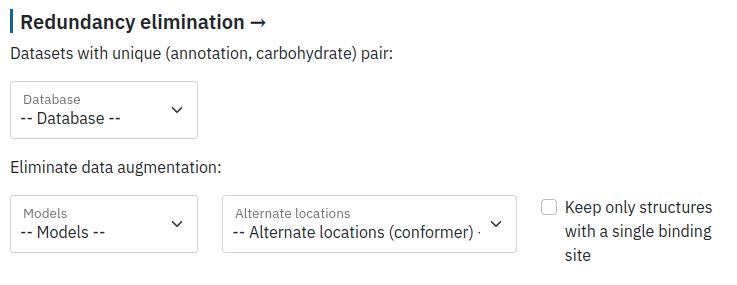
Eliminate redundancy
Finally, the user can filter redundancy in their data by keeping a unique ID according to different databases and selecting a unique model from NMR structures.
Compare sugar binding sites
For a given protein-carbohydrate complex, the user has a possibility to retrieve all the similar protein-carbohydrate interfaces found in DIONYSUS. The implemented algorithm is using a graph-based score to compare the geometry and composition of two carbohydrate binding sites (see About for more details).
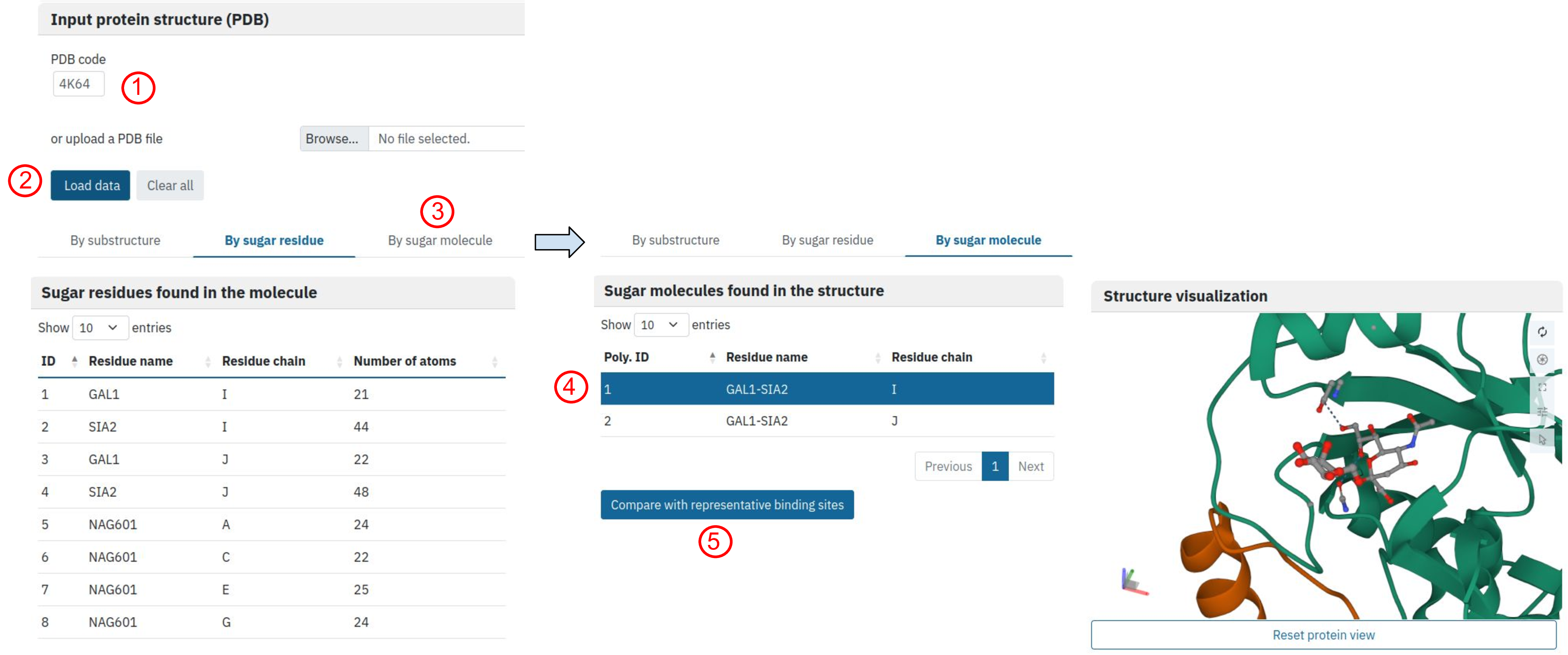
The user can upload a protein-carbohydrate complex of interest either using a PDB ID or directly from file. Here we explore carbohydrate binding site found on the surface of avian influenza H5 hemagglutinin, which was complexed with analog of human receptor (PDB ID: 4K64)
Once the file is uploaded (click 1 then 2), the automated script selects all the carbohydrate-containing residues. For each residue we indicate the number of atoms on the protein surface in 7A radius from the atom of the carbohydrate ring. The user can either select one of the carbohydrate residues (“By sugar residue option”) or search all binding sites associated with the sugar ligand (“By sugar molecule”, click 3). We have chosen the second option, and decided to select and visualise the first ligand found in the structure (click 4) and search for similar binding sites in our database (click 5).
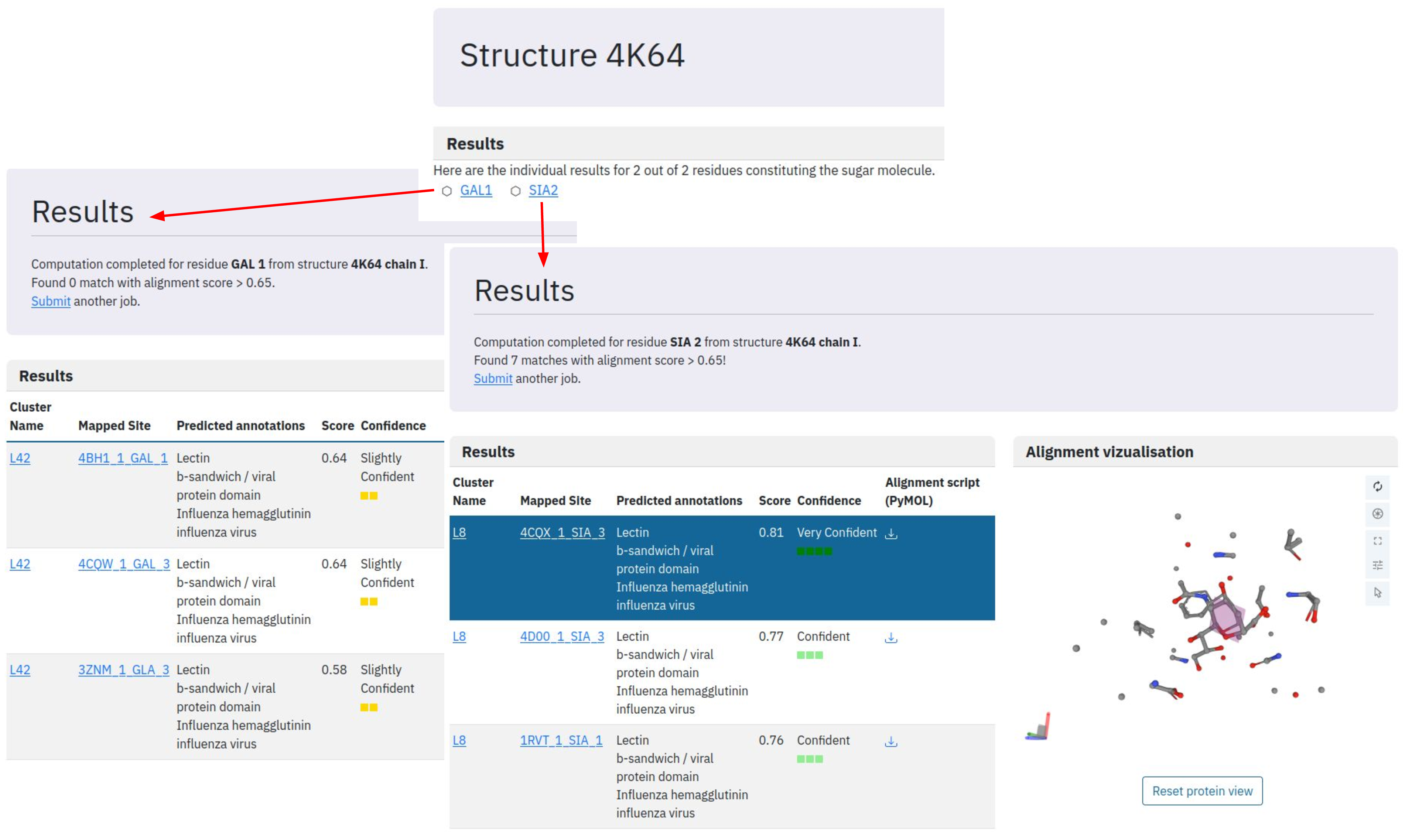
Once the job is submitted, the algorithm launches a comparison for each residue against the representative dataset of binding sites in the DIONYSUS database and reports corresponding matches with confidence level. We consider the predicted mapping reliable for score above 0.65 and highly reliable for score above 0.8. In our example, we do not find any similar galactose binding sites (all the matches have a score below 0.65). At the same time, we find several structures with highly similar binding site patterns for sialic acid residue. All the similar binding sites belong to lectin cluster L8 and the most resembling one is found in the structure with PDB ID: 4CQX. The binding site superposition is shown upon a click on the corresponding binding site but further detailed visualisation can be done using PyMOL alignment script provided for download.
To produce visualisation in PyMOL, the user should open PyMOL and run the alignment script. In our case the command will look like:
run run_DIONYSUS_downloaded_files_Em43khWnhQ-tables_cluster_L8-9.pyThe script will fetch all the necessary structure files, align them according to the carbohydrate binding site superposition and zoom to the binding site.
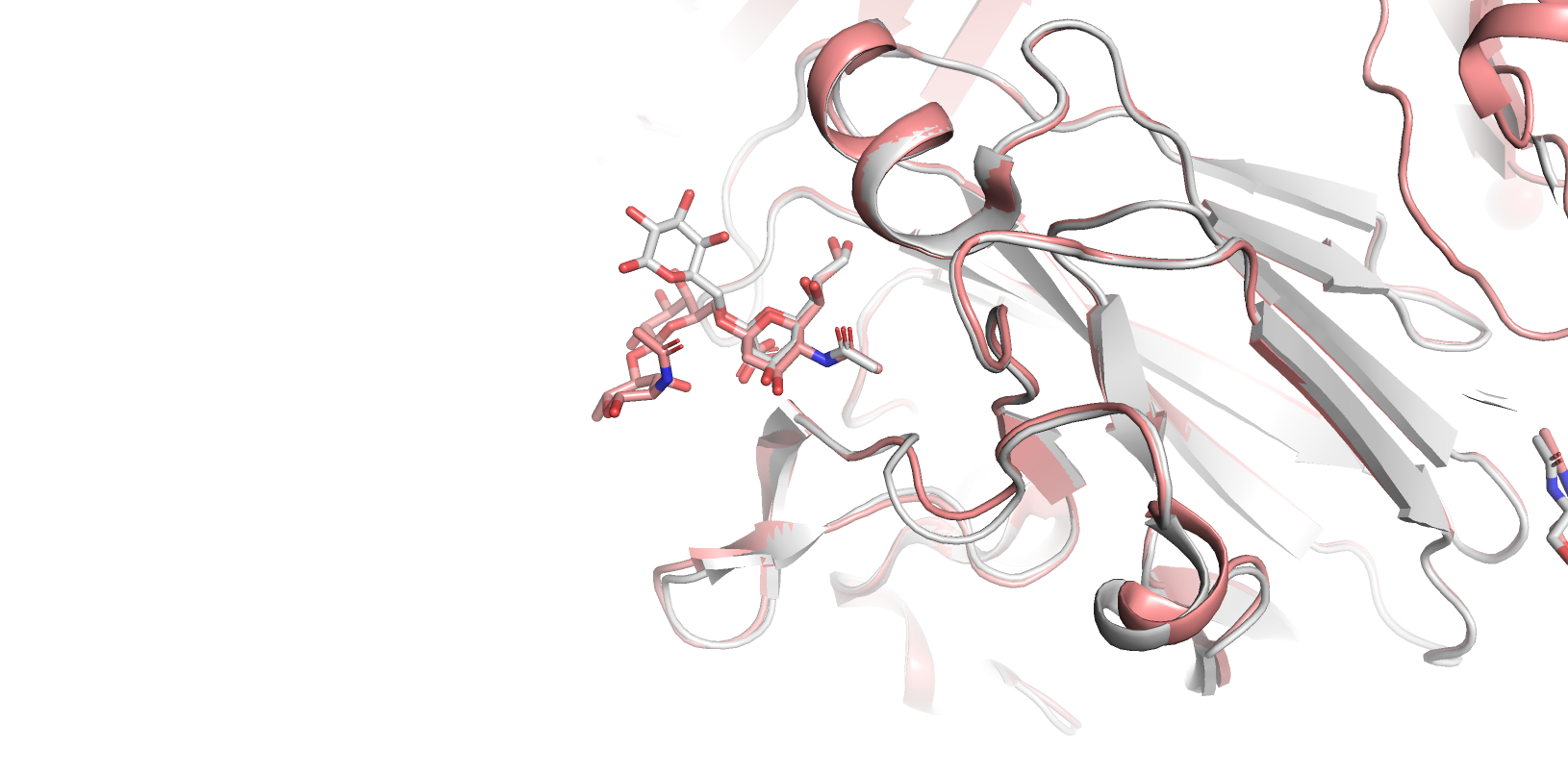
In our case, protein alignment confirms almost identical organisation of carbohydrate binding sites in our target protein and in Influenza hemagglutinin. Interestingly, our target protein is not yet repertoire in the UniLectin database and therefore our results provide probable classification for this protein: cluster L8.
Explore clusters of sugar binding sites
We provide a t-SNE projection of different clusters found in lectins, in carbohydrate-active enzymes of different classes (full annotations can be addressed by clicking on “Show legend button”), antibodies and carbohydrate binding modules (CBMs). Each symbol represents the most common carbohydrate (according to SNFG nomenclature) and the size of each symbol is proportional to the number of different proteins in the same cluster.
The user can select proteins for comparison by checking the corresponding boxes. For example, below we select only clusters of lectin binding sites. Then, by double-clicking on the sialic-acid symbol, we further restrain our selection to clusters with prevalence of the sialic acid residue as carbohydrate ligand.
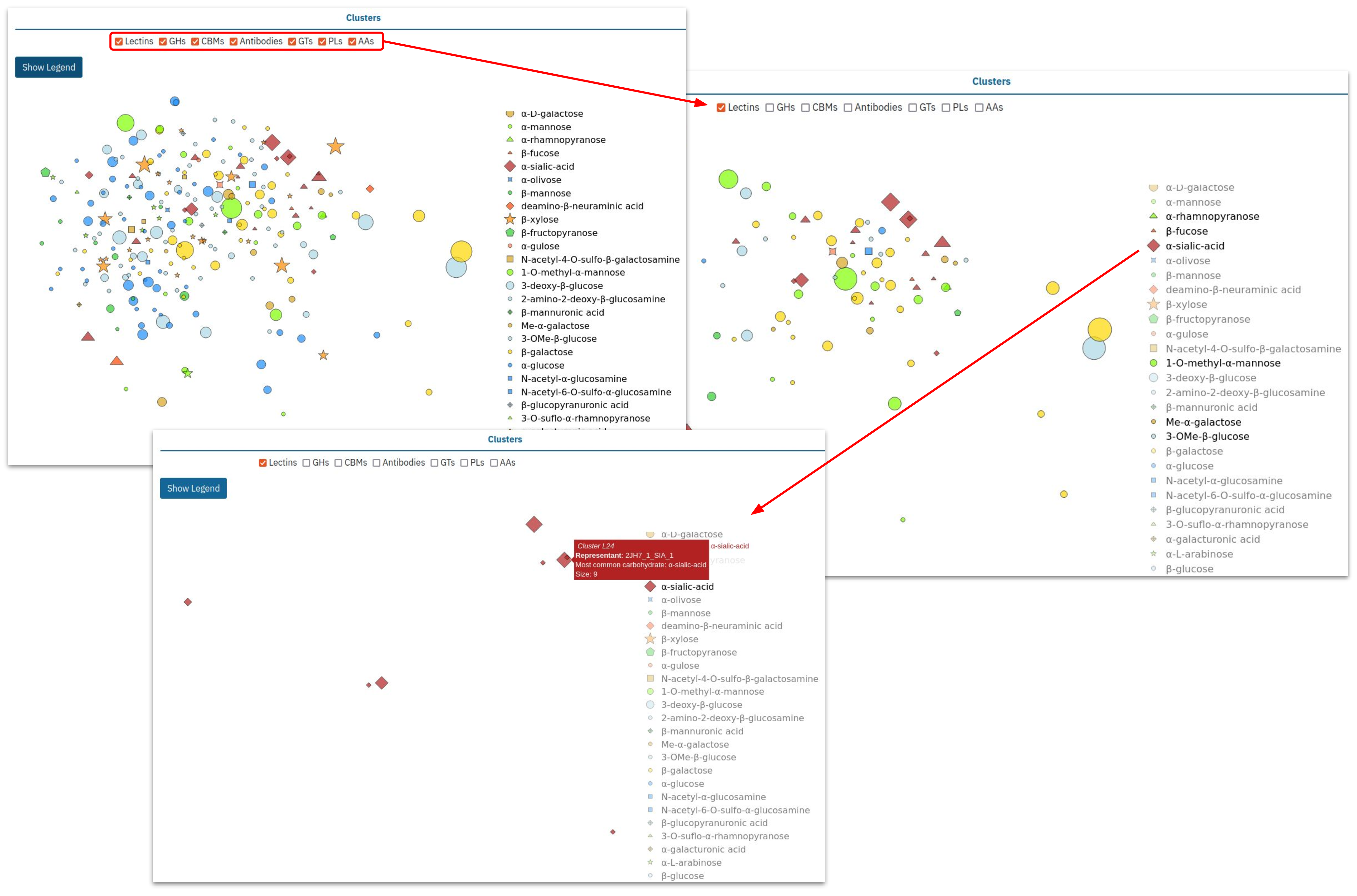
Among the selected clusters, L24 attracts our attention as one of the most diverse. The interactive diagram allows us to directly access its page: L24.

The page header provides a summary of cluster content. We report common protein annotations according to UniLectin, CAZy, SabDab, their fold according to ECOD and their functions according to UniProt as well as most common carbohydrate residues involved in the interaction.
The user can also explore cluster content by analyzing interactive graphs describing three main aspects: carbohydrate properties, binding site properties, and data reliability.
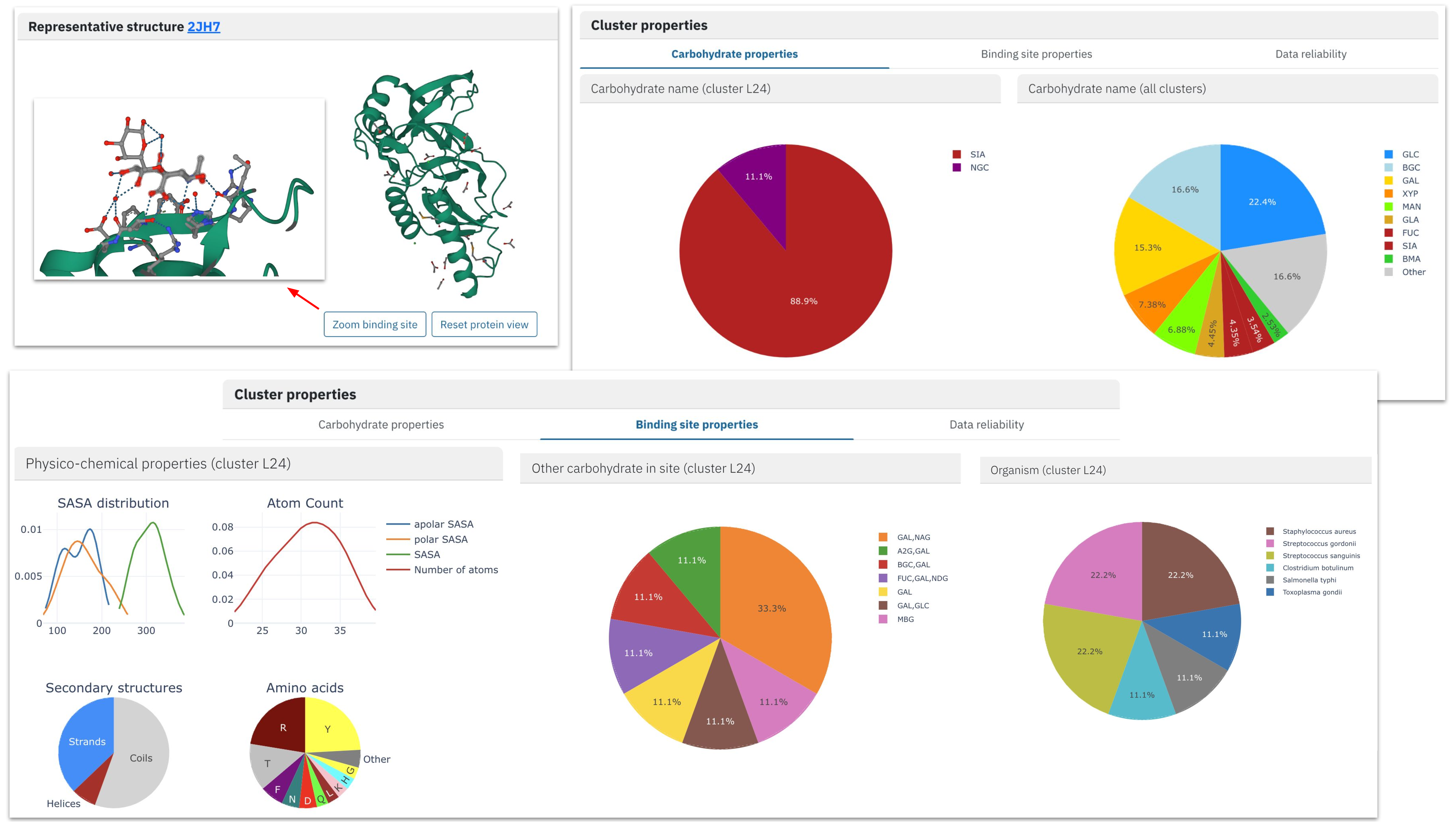
Carbohydrate content of our cluster is very homogeneous, and the absolute majority of binding sites are formed by sialic acid. At the same time, we find other monosaccharide molecules in the proximity of sialic acid residues, forming di- and oligosaccharide ligands.
Analysis of the binding site properties confirms the diversity of proteins gathered in the same cluster: they come from six different proteins from different kingdoms of life. While the protein architectures are also very different (with various ECOD classes), the majority of binding sites are formed by beta-strands regions of the protein (“Physico-chemical properties”) with characteristic amino acid content. The characteristic local arrangement is also visible on the representative binding site structure.
Finally, at the bottom of the page, we provide an interactive table with all the binding sites. The user can further refine the selection by choosing binding sites:
- of high-resolution quality (without detected artefacts such as broken ligand structure or atomic clashes)
- classified as biologically relevant according to BioLip
- with available binding affinity
- corresponding to active sites according to UniProt
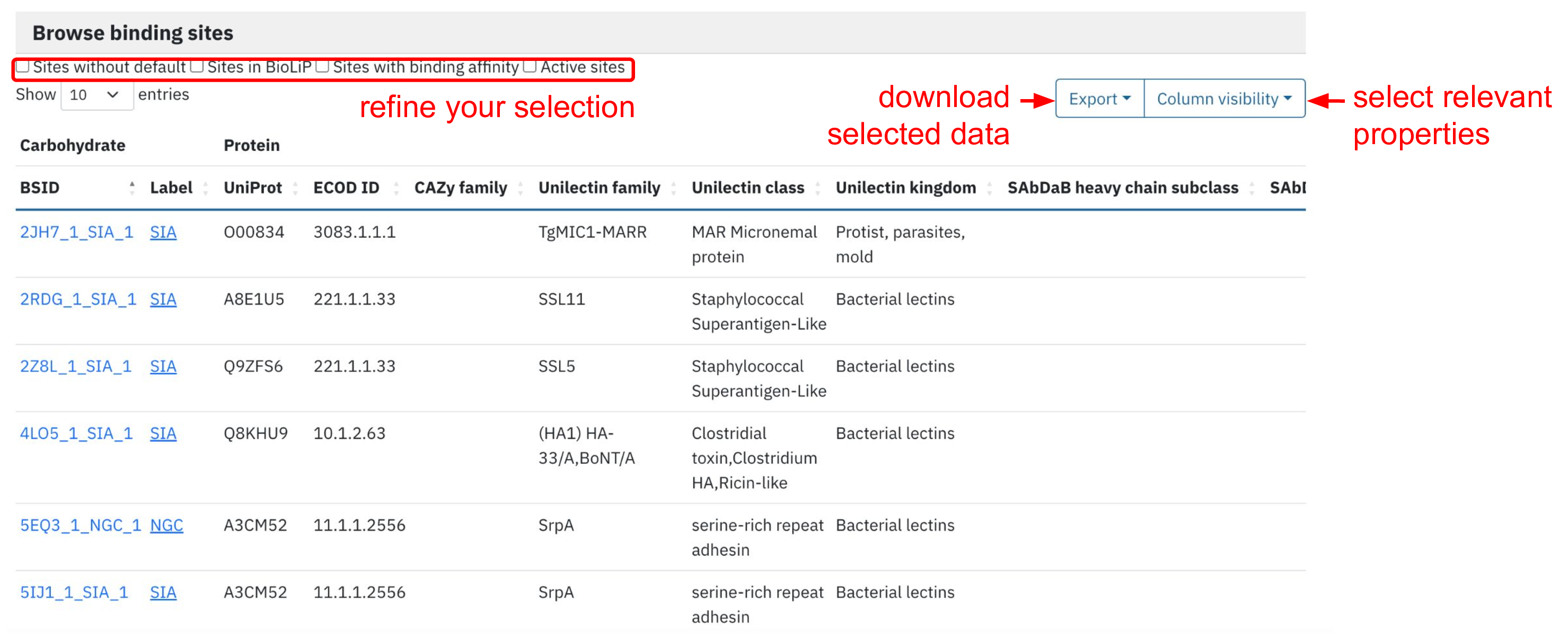
The user can also add/remove protein annotations using “Column visibility” button. The resulting table can be exported in the convenient format such as .csv.
Analyze glycosylations
For each experimental structure, we have identified different types of glycosylations according to the distance cutoff between the protein and carbohydrate atoms and coupled it to the information available in UniProt. Each color indicates a glycosylation type: red for O-glycosylation, blue for N-glycosylation, and gray for C-glycosylations.
Undetermined types of interactions are given in black and are reported in the following cases:
- Chemical reaction between carbohydrate ligand (see for example, BS1 of 2GDV)
- S-glycosylation (e.g. BS2 of 2KUY)
- Resolution artifact (e.g. BS11 of 6TVF, N-Acetylglucosamine is bound to the protein via a C-O bond instead of the usual C-N bond)